Šis klīniskais gadījums vēl nav līdz galam atrisināts, tomēr tas ir pietiekami interesants, lai par to rakstītu. Šajā gadījumā ir klīniskas un anamnestiskas aizdomas par salīdzinoši retu ģenētisku saslimšanu, iespējams, nepietiekami bieži diagnosticētu.
Klīniskā gadījuma demonstrācija
(dr. Lauma Zariņa)
Paciente ir 23 gadus veca sieviete. 2010. gada 12. aprīlī vērsusies P. Stradiņa Klīniskās universitātes slimnīcas Neatliekamās medicīnas centrā (NMC) ar sūdzībām par nespēku, sirdsklauvēm, bezmiegu. Šādas sūdzības ir pēdējās divas nedēļas, tās dinamikā pastiprinās.
Ģimenes anamnēze
Pacientes māte mirusi 41 gada vecumā. Pēc radinieku teiktā, viņai bijušas recidivējošas hemangioblastomas galvas smadzenēs un veidojumi abās virsnierēs. Tikušas veiktas atkārtotas hemangioblastomu rezekcijas, kā arī esot veikta vienpusēja adrenalektomija. Pacientes mātei bijušas arī hipertensīvās krīzes. Paciente ir vienīgais bērns ģimenē.
Slimības anamnēze
Pacientei ir bijusi viena grūtniecība, kas rezultējusies ar dzemdībām 2007. gadā. Tika veikts ķeizargrieziens, iemesls - nepareiza augļa guļa. Grūtniecības norise bijusi bez starpgadījumiem.
2008. gadā pacientei ir bijusi ģībšanas epizode, pēc kuras tika veikts MR izmeklējums galvas smadzenēm un tika konstatēts veidojums labās puses deniņu daivā. Pacientei tika veikta labās puses deniņu daivas hemangioblastomas operācija.
Ņemot vērā šo klīnisko atradi (hemangioblastoma) un ģimenes anamnēzi, 2008. gadā, izrakstot pacienti no slimnīcas "Gaiļezers", tiek izteiktas aizdomas par Hipela-Lindava (von Hippel-Lindau) slimību un rekomendēts veikt datortomogrāfiju vēdera dobumam. Izmeklējums tiek izdarīts 2008. gadā Valmieras slimnīcā, konstatēts 3-3,5 cm liels veidojums kreisajā virsnierē, taču nekādi citi izmeklējumi nav veikti.
Līdz šā gada aprīlim paciente it kā dzīvo normālu dzīvi, nekādu medikamentozu terapiju nelieto, hipertensīvas krīzes nav bijušas. Vidēji arteriālais asinsspiediens 110/60 mmHg. Alerģijas paciente noliedz, medikamentus regulāri nelieto.
Objektīvais stāvoklis NMC
Paciente ir astēniska, svars 48 kg. Āda pabāla, mikla. Sirdsdarbība ritmiska, 100 x min., arteriālais asinsspiediens 110/70 mmHg. Plaušās vezikulāra elpošana, 18 x minūtē. Ķermeņa temperatūra 36,8ºC. Vēders mīksts, nesāpīgs. Perifēru tūsku nav.
Analīzes slimnīcas NMC: pilna asinsaina un asins bioķīmija normas robežās (hlorīdi: 105 []; nātrijs: 139 []; kālijs: 4,8 []; kreatinīns: 49 []; glikoze: 6,5 []; AsAT: 16 []; AlAT: 14 []).
Ņemot vērā pacientes slikto pašsajūtu, ģimenes anamnēzi un to, ka paciente līdz galam nav izmeklēta, viņu stacionē 9. nodaļā tālākai izmeklēšanai.
Izmeklēšana 9. nodaļā
DT vēdera dobumam: labā virsniere neizmainīta. Kreiso virsnieri aizņem ap 4,0 cm liels, apaļš, labi norobežots veidojums, kura blīvums natīvā 30 HV. Pēc k/v ievades redzams, ka tas ir krasi hipervaskulārs, vaskularizācija neviendabīga, blīvums kopumā pieaug līdz 170 HV. 15 min. laikā notiek k/v izskalošanās, blīvumam samazinoties līdz 70 HV. Citur vēderā patoloģiskus veidojumus neatrod.
Slēdziens: veidojums kreisajā virsnierē, jādomā, benignas dabas (k/v izskalošanās virs 50%).
Kateholamīni diennakts urīnā: adrenalīns 15,5 mkg/24 h (4-20), dopamīns 421 mkg/24 h (190-450). Tomēr pēc šīs atrades vien, ņemot vērā Hipela-Lindava slimības klīniskās pazīmes un ģimenes anamnēzi, nevaram izslēgt, ka veidojums ir feohromocitoma snaudošā stadijā, kas pie noteiktiem palaidējmehānismiem var manifestēties ar masīvu kateholamīnu izdali.
MR galvai: osteoplastiskas trepanācijas lēvers labā deniņa/paura robežrajonā pēc hemangioblastomas operācijas. Smadzeņu vēderiņi normas robežās, nedaudz platāks labā sānu vēderiņa pakauša rags. Pārliecinoši norādījumi par reziduāliem tumora audiem vai prolongētu augšanu nav iegūti. Citur smadzeņu struktūrā patoloģiskas pārmaiņas nenovēro. Intrakraniālās artērijas un to zari bez patoloģiskām pārmaiņām vai īpatnībām.
DT plaušām (lai izslēgtu hemangiomas plaušās): pārliecinošus norādījumus par patoloģiju neiegūst.
Okulista konsultācija: tiek pārbaudīta tīklene, oftalmoloģiski paciente ir vesela.
Endokrīnās ķirurģijas konsilijs: endokrinologi, ķirurgi un radiologi
(2010. gada 16. aprīlī)
Ņemot vērā slimības un ģimenes anamnēzi:
- indicēts veikt ģenētisko izmeklēšanu, jo ir aizdomas par ģenētisko sindromu;
- indicēta ķirurģiska terapija. Tās pamatindikācijas- nevar izslēgt, ka šis veidojums ir feohromocitoma; veidojums ir liels (4cm) un salīdzinoši dinamikā palielinās. Lai arī patlaban DT tas tiek aprakstīts kā benignas dabas, nevar izslēgt malignitātes attīstību;
- nodrošināt adekvātu sagatavošanu ķirurģiskai terapijai ar alfa adrenoblokatoriem 1mēnesi- T.Cardura 1mgx1 vakaros.
Ģenētisko analīžu izmeklējuma slēdziens: Hipela-Lindava audzēja supresora proteīna 157. pozīcijā treonīna vietā ir izoleicīns (T157I), kas apstiprina Hipela-Lindava slimību.
Diagnoze
- Hipela-Lindava slimība, IIA tips.
- Kreisās virsnieres jaunveidojums, feohromocitoma suspecta.
- Stāvoklis pēc labās puses deniņu paura daivas hemangioblastomas rezekcijas 2008.gadā.
2010. gada 16. aprīlī paciente tiek izrak-stīta tālākai ambulatorai terapijai, ambulatori tiek nozīmēta audiogrammas veikšana.
Atkārtota stacionēšana
(2010. gada 6. maijā)
Sūdzības: spiedoša sajūta pakauša rajonā, reiboņi, apetītes trūkums, nespēks, subfebrīla temperatūra aptuveni 3 nedēļas.
Objektīvi: arteriālais asinsspiediens 110/75 mmHg, tahikardija 120 x min. Temperatūra 37,5ºC. Nodaļā ir veikti izmeklējumi, kur objektīvus sūdzību izraisošus cēloņus neatrod. Iekaisuma marķieri ir negatīvi. EhoKG, doplerogrāfija brahiocefālajiem asinsvadiem - bez novirzes.
Neirologa konsultācija - rekomendē psihiatra konsultāciju, jo atzīmē, ka prevalē depresīvais sindroms.
Pacientei tiek sākta antidepresantu terapija.
Diskusija
Prof. A. Danilāns: Pacientes svars ir 48 kg. Vai sen jau tas ir tāds vai pēdējā laikā?
Dr. L. Zariņa: Viņai svars pastāvīgi bijis ap 50 kg, bet pēdējā laikā ir nedaudz samazinājies.
Prof. A. Pētersons: Vai pacientei ir jebkad konstatēts paaugstināts asinsspiediens?
Dr. L. Zariņa: Nē, nav. Viņa pati gan atzīmē, ka vienu reizi bijis 130/70 mmHg, bet bez hipertensīvās krīzes klīniskajām pazīmēm.
Dr. G. Geldnere: Tahikardija, kā es sapratu, viņai bijusi vienmēr?
Dr. L. Zariņa: Faktiski jā. Bet pēc pirmās stacionēšanas, uzzinājusi izmeklēšanas rezultātus un sīkāk par slimību, kā arī, ņemot vērā ģimenes anamnēzi, paciente kļuva emocionāli ļoti satraukta - bija izteiktāka tahikardija, subfebrīla temperatūra.
Dr. G. Geldnere: Vai beta blokatori viņai nevarēja palīdzēt?
Dr. L. Zariņa: Ņemot vērā pamatotas klīniskas aizdomas par feohromocitomu, beta blokatorus sākotnēji nozīmēt nedrīkstējām. Tie ārstēšanā tika pievienoti vēlāk, kad paciente jau saņēma alfa blokatoru terapiju.
Prof. V. Pīrāgs: Vai ievācāt arī plašāku ģimenes anamnēzi? Vai šajā ģimenē ir zināmi neskaidri nāves gadījumi? Šis faktiski ir iedzimtā vēža, iedzimto audzēju sindroms, un tas ir sastopams biežāk, nekā mēs domājam.
Dr. L. Zariņa: Protams, mums ir interese šo gadījumu izpētīt daudz rūpīgāk. Ir runāts arī ar ģimeni, plānots ģenētiskās analīzes veikt arī radiniekiem. Šobrīd gan nav norādes, vai kādam no radiniekiem varētu būt šis sindroms. Atskaites punkts ir pacientes vecvecāki. Viens no viņiem varētu būt šā gēna nesējs, bet faktiski ne vienam, ne otram nav bijušas izteiktas hipertensīvās krīzes. Neskaidrs nāves gadījums ģimenē ir bijis viens - kāds ļoti attāls radinieks.
Prof. V. Pīrāgs: Apmēram reizi divos gados Latvijā piedzimst viens šāds jaundzimušais. Droši vien diagnosticēts tiek daudz mazāk pacientu.
Prof. A. Pētersons: Pacientes vienīgā iespēja, manā izpratnē, ir nodzīvot tik ilgi kā viņas mātei un vēl ilgāk. Un tas ir iespējams tikai un vienīgi tad, ja "dzenas" pakaļ šiem atsevišķajiem tumoriem un tos eliminē. Varbūt šajā konkrētajā gadījumā mūs mazāk uztrauc feohromocitomas aktivitāte, kas, šķiet, šobrīd nav īpaši izteikta, bet vairāk tas, ka tumors var būt maligns. Bez operācijas to pateikt nav iespējams.
Teorētiskā daļa
(dr. Jekaterina Kovzeļa)
Hipela-Lindava slimība (von Hippel-Lindau (VHL)) slimība ir AD pārmantojams sindroms, kas izpaužas ar dažādiem labdabīgiem un ļaundabīgiem tumoriem. VHL gēna anomālija ir apmēram vienam no 36 000 jaundzimušo. Slimības manifestācija parasti ir apmēram 26 gadu vecumā, bet var manifestēties arī bērnībā, pusaudžu vai pieaugušo vecumā.
Vēsture
Nepilnīgi šīs slimības apraksti bija zināmi jau pirms vairāk nekā 100 gadiem. 1904. gadā vācu oftalmologs Eugen von Hippel aprakstīja retinālas angiomas ģimenes locekļiem vairākās paaudzēs. 1926. gadā zviedru patologs Arvid Lindau publicēja disertāciju, kur aprakstīja retinālas angiomas, smadzenīšu hemangioblastomas, nieru, aizkuņģa dziedzera, sēklinieka piedēkļa cistas kā daļu no pārmantota sindroma. 1964. gadā publicēti pirmie diagnozes kritēriji, t.sk. nefrokarcinoma. 1988. gadā šo slimību sasaistīja ar 3. hromosomu. 1993. gadā Latif un kolēģi identificēja VHL tumora supresora gēnu.
VHL klasifikācija
- I tips- VHL bez feohromocitomas.
- II tips- VHL ar feohromocitomu:
- IIA tips. Feohromocitomas, retinālas angiomas, CNS hemangioblastomas; zema nefrokarcinomas incidence;
- IIB tips. Feohromocitomas, retinālas angiomas, CNS hemangioblastomas, pancreas neoplazmas un cistas; augsta nefrokarcinomas incidence;
- IIC tips. Tikai feohromocitoma.
VHL molekulārā bioloģija un patoģenēze
VHL gēns atrodas 3p25 hromosomā. Mutācijas tiek atrastas VHL gēna visos (trīs) eksonos un dažos intronos. Mutāciju tipi: misens mutācijas, nonsens mutācijas, daļējas un pilnīgas gēna delēcijas. 2004. gadā bija zināms jau vairāk nekā 500 mutāciju, šobrīd varētu būt jau 1500.
Gēna produkts ir proteīns pVHL, kas kalpo kā tumora supresors. pVHL sastāvā ir a-domēns, kam piesaistās elongīns un citi proteīni, veidojot stabilu kompleksu. Šis komplekss funkcionē kā receptors mērķa molekulām. b-domēns ir domāts šo mērķa molekulu piesaistīšanai, piemēram, HIF-1. pVHL veicina šo mērķa proteīnu protesomālo degradāciju, tādējādi regulējot to līmeni šūnā. Otrā svarīgā nozīme VHL patoģenēzē ir HIF-1 molekulai. Hipoksijas in du cē tais faktors (HIF-1) ir viens no galvenajiem proteīniem, ko regulē pVHL. HIF-1 sastāv no a un b subvienībām. HIF-1 a subvienība ir jutīga pret O2 līmeni un ir pVHL kompleksa substrāts. pVHL saistās ar elongīnu C, elongīnu B, Cul2, RBX1, veidojot multimērisku kompleksu. Šis komplekss degradē HIF-1 a. Normāli pie laba O2 piesātinājuma notiek HIF-1 a degradācija. Hipoksijas apstākļos HIF-1 netiek degradēts un tas pavairojas. VHL gēna mutācija un defektīvs pVHL paaugstina HIF-1 līmeni. Tādējādi VHL slimības modelis var atgādināt hipoksijas situāciju, lai gan šūnā ir normāls O2 parciālais spiediens.
Paaugstināts HIF-1 līmenis ir saistīts ar palielinātu vairāku gēnu transkripciju: vaskulāra endotēlija augšanas faktors (VEGF), eritropoetīns, PDGF, Glut1, transformējošais augšanas faktors-a, matrices metalloproteināzes, atipiska proteīnkināze C. VEGF ir zināms kā promoters tumoru angioģenēzei, kas pamato VHL tumoru vaskulāro dabu.
Ar VHL asociētie tumori
Hemangioblastomas
Tās ir labi norobežotas, labi vaskularizētas labdabīgas neoplazmas (skat. 1. attēlu). Var izraisīt simptomus blakus audu saspieduma vai asiņošanas dēļ. Tās ir visbiežākā VHL izpausme 60-84% pacientu. Atšķirībā no sporādiskām hemangioblastomām VHL gadījumā tās ir multiplas. 51% no tām ir lokalizētas spinālā kanālā, 38% smadzenītēs, 10% smadzeņu stumbrā, 2% smadzeņu puslodēs. Hemangioblastomas var palikt "snaudošas" ilgu laiku vai var ātri augt. Mazus un asimptomātiskus veidojumus iespējams novērot. No ķirurģijas atturas, kamēr veidojumi kļūst simptomātiski vai ātri aug. Stereotaktiskā radioķirurģija un konvencionāla frakcionēta staru terapija ārstē grūti sasniedzamos veidojumus un ļau
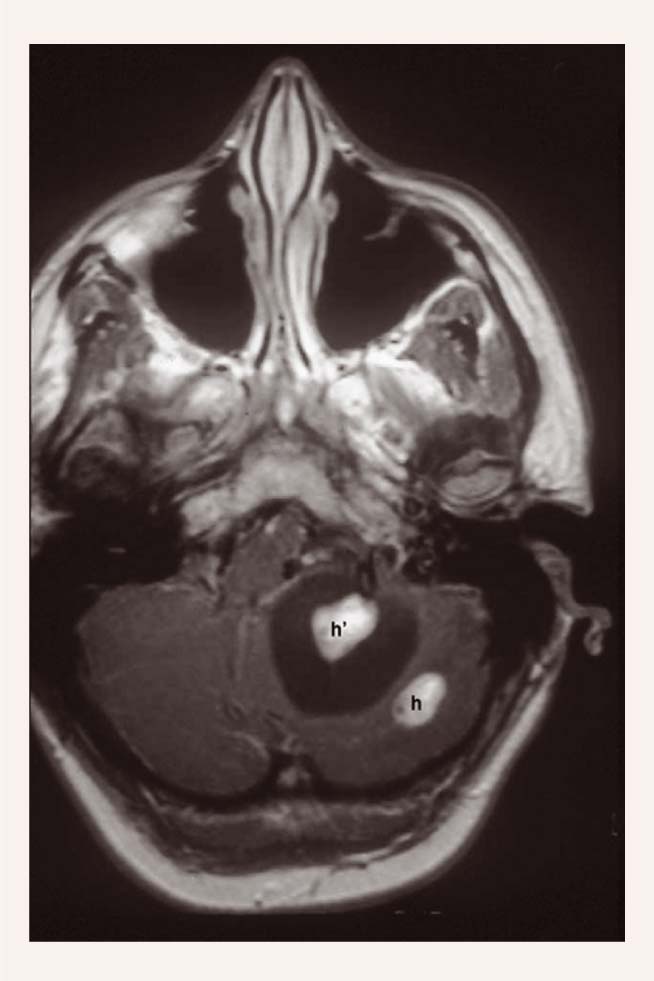
1. attēls
Hemangioblastoma
j izvairīties no biežām operācijām. Nav pētījumu, kas parāda atšķirību efektivitātē un drošībā.
Retinālās angiomas
Retinālās angiomas ir hemangiobastomas retīnā un redzes nervā. Ja neārstē, var asiņot, novest pie tīklenes atslāņošanās, glaukomas un redzes zuduma. Atrod apmēram 70% VHL pacientu, parasti multifokālas un bilaterālas. Salīdzinot ar sporādiskām angiomām, VHL pacientiem tās atrod jaunākā vecumā un tās ir multiplas. Pacientam atrodot retinālas angiomas, tās rekomendē ārstēt uzreiz, lai novērstu augšanu un komplikācijas. Lāzera fotokoagulācija un krioterapija efektīva 70% gadījumu, tā ir izvēles metode. Izņēmums ir redzes nervs - ar šīm metodēm to nedrīkst ārstēt blakusparādību dēļ. Šādos gadījumos var izmantot ārējo staru terapiju.
Gaiššūnu nefrokarcinoma
Divām trešdaļām pacientu attīstās multiplas renālas cistas un karcinoma. Visas VHL karcinomas ir gaišo šūnu tumori (skat. 2. attēlu). Nepilni 25% no tumora var saturēt papillāru komponentu. Ja dominē papillāra, hromofoba, onkocistiska histoloģija, tad tumors nav saistīts ar VHL slimību. Reti ir pacientiem, kas jaunāki par 20 gadiem. Karcinoma parasti ir multicentriska un bilaterāla, izceļas vai nu saistībā ar nieru cistām, vai de novo no necistiskās nieru parenhīmas. Kaut gan nieru cistas var būt labdabīgas, tās tiek uzskatītas par pirmsvēža stāvokli. Cista ar blīvu saturu, kas izskatās kā labdabīga, gandrīz vienmēr satur nefrokarcinomu. Veselas nieru parenhīmas histopatoloģiska izmeklēšana parāda dažādas gaiššūnu anomālijas, kas arī uzskatāmas par nefrokarcinomas prekursoriem. Šādus gaišo šūnu prekursorus neatrod pacientiem ar sporādisku nefrokarcinomu vai veseliem cilvēkiem.
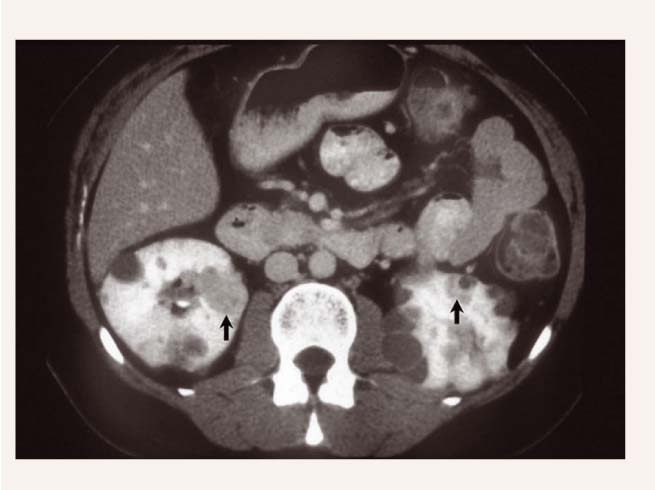
2. attēls
Gaiššūnu nefrokarcinoma
Terapija - no radikālas nefrektomijas līdz nefronus saudzējošai terapijai (mazu bojājumu novērošana, daļēja nefrektomija, krioterapija, radiofrekvences ablācija). Piemēram, tumoriem zem 3 cm ir zema metastazēšanās spēja, tāpēc tos rūpīgi novēro. Pēc terapijas turpināma rūpīga novērošana, 5 gadu laikā jaunas tumora masas atrodamas 30% pacientu, pēc 10 gadiem - 85%. Metastātiskas slimības risks ir zems, ja pacientu rūpīgi monitorē. Nieru transplantācija tiek veikta pacientiem, kam indicēta bilaterāla nefrektomija vai attīstās termināla nieru mazspēja. Bet to ierobežo tas, ka imūnsupresīva terapija paaugstina tumora recidīva risku.
Feohromocitoma
Tikai VHL II tipa slimnieku radiniekiem ir liels feohromocitomas (skat. 3. attēlu) risks. Bet, tā kā šī korelācija ir nepilna, visi pacienti ar VHL mutāciju tiek novēroti. Ar VHL slimību saistīta feohromocitoma tiek atrasta jauniem pacientiem, bieži ir multipla un ekstraadrenāla, tā retāk dod simptomus vai kateholamīnu pārprodukciju, salīdzinot ar slimniekiem bez VHL. Nereti novēroti arī gadījumi pediatrijā. Vie nā pētī ju mā no 64 VHL pacientiem 12% feohromacitoma bija ekstraadrenāla, 35% asimptomātiska. [1] Tumoriem, kas producē kateholamīnus, ir ti piska klīniskā aina. Bet asimptomātiskas feohromocitomas iespēja ir jāapsver, ja šiem pacientiem nepieciešama ķirurģiska ārstēšana, jo ir paaugstināts risks uz simpatisku hiperaktivitāti un smagu hipertensiju.
Ārstēšana ir ķirurģiska. Parciāla adrenalektomija un enukleācija tiek rekomendēta, lai saglabātu virsnieres funkciju. Pirms operācijas pacients jāsagatavo ar alfa-blokatoriem, vēlāk pievienojot beta-blokatorus.
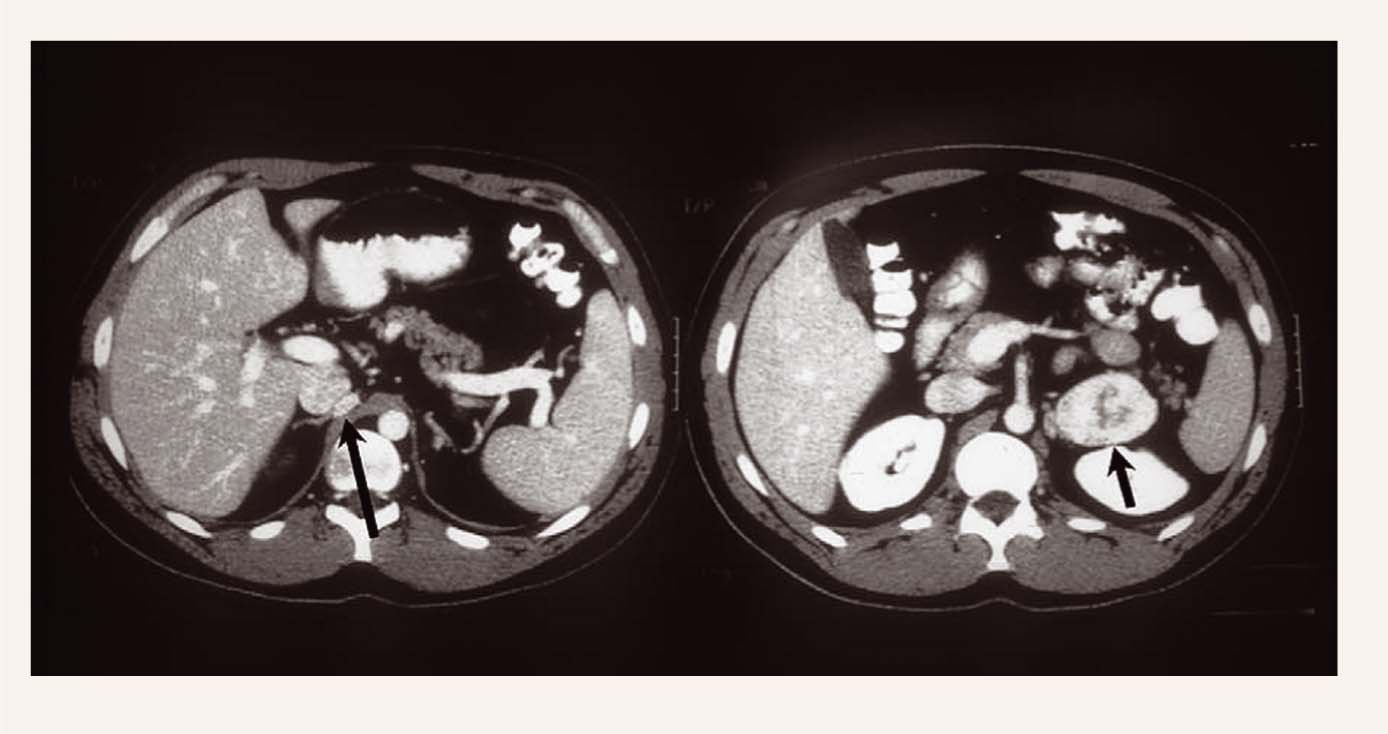
3. attēls
Feohromocitoma
Endolifātiskā maisa tumori vidusausī
Papillāras cistadenomas vidusauss endolimfātiskajā maisā ir augsti vaskularizēti bojājumi, veidojas os temporalis posterior iekšā (skat. 4. attēlu).
Manifestējas kā dzirdes zudums, tinnīts, vertigo, retāk - kā sejas muskuļu vājums.
Lai gan šie tumori mēdz būt sporādiski, VHL slimniekiem tie attīstās salīdzinoši agrīnā vecumā un bieži ir bilaterāli. VHL pacientiem, kam ir dzirdes vai vestibulārie simptomi vai izmaiņas audiogrammā, jāveic DT vai MRI.
Ārstēšana ir ķirurģiska. Stereotaktisko radioķirurģiju var izmantot slimības recidīvos.
Kurluma gadījumā var izmantot kohleāro implantu.
Aizkuņģa dziedzera tumori
Neiroendokrīnie pancreas tumori var metastazēt aknās un izraisīt simptomus atbilstīgi izdalītajiem peptīdiem (piemēram, VIP - diareja, insulīns - hipoglikēmija utt.). Lielākā daļa šo tumoru ir asimptomātiska un aug ļoti lēni bez simptomiem, kas varētu būt peptīdu pārprodukcijas dēļ. Tāpēc bieži šos tumorus atrod nejauši.
Neiroendokrīno tumoru ārstēšana ir ķirurģiska. Rezekciju veic, ja tumors pārsniedz 2 cm pancreas galviņā vai 3 cm pancreas ķermenī vai astē. Mazākus asimptomātiskus tumorus var novērot reizi gadā.
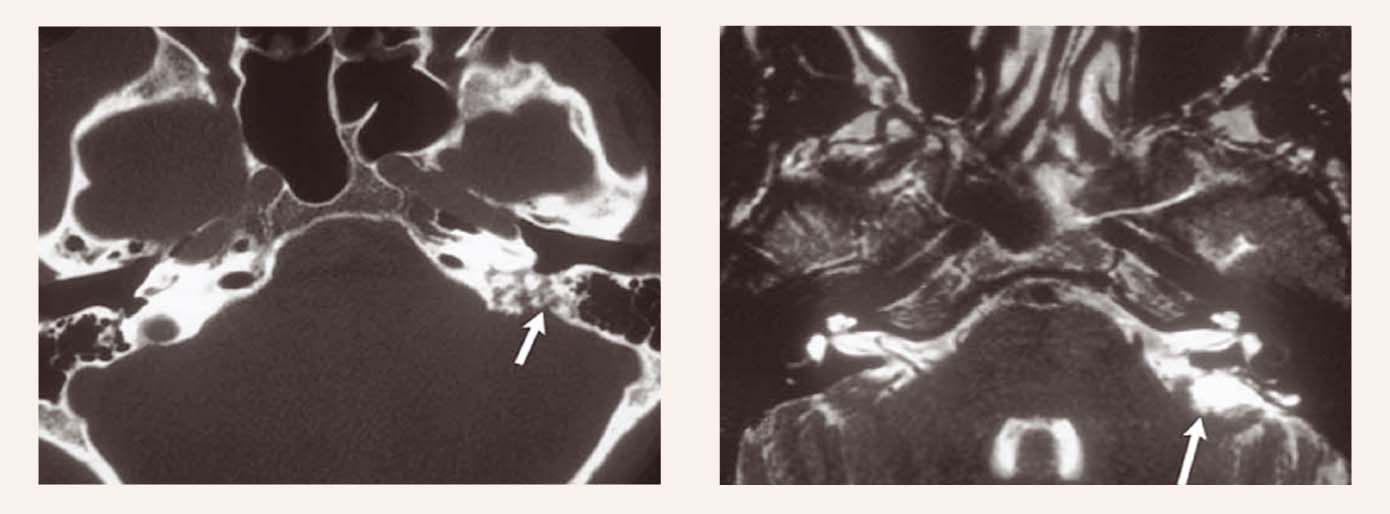
4. attēls
Endolifātiskā maisa tumori vidusausī
Epididymis un lig. latum uteri papillāras cistadenomas
Papillāras cistadenomas attīstās vīriešiem sēklinieka piedēklī un sievietēm dzemdes platajā saitē. Atsevišķas sēklinieka piedēkļa cistas vispārējā populācijā bieži sastopamas, un par VHL aizdomām nevajadzētu būt, ja nav citas atrades. No otras puses, bilaterālas cistas ir patognomas VHL slimībai.
Epididimālas cistas ir labdabīgas un parasti asimptomātiskas, terapija nav nepieciešama. Dzemdes platās saites papillāras cistadenomas arī ir asimptomātiskas lielākajai daļai pacienšu.
Iespējamie simptomi: sāpes, dyspareunia, menorāģija. Ārstēšana simptomātiska.
Diagnostika
Diagnozes apstiprināšanai izmanto ģenētiskos testus. Ģenētiskajai analīzei jāpaņem asins paraugs EDTA stobriņā. VHL gēna mutācija tiek meklēta limfocītos. Bērns, kas piedzimis ģimenē ar zināmu VHL slimību, tiek izmeklēts uz to pašu mutāciju ar Sauzerna blot-hibridizācijas metodi. Ja ir aizdomas par VHL slimību, bet ģimenes anamnēzē VHL mutācija nav zināma, ir jāmeklē punktveida mutācijas, sekvencējot visu VHL gēnu.
Testa sensitivitāte un specifitāte tuva 100%. To, vai pēcnācējs pārmantojis mutāciju, var uzzināt pēc viņa piedzimšanas vai prenatāli ar amniocentēzes palīdzību, vai izmeklējot horija bārkstiņas. Topošie vecāki jāinformē par reproduktīvām tehnoloģijām, kas mazina risku pēcnācējiem saslimt ar VHL, piemēram, donoru sperma vai oocīts (atbilstīgi tam, kurš no vecākiem ir slims) un ģenētiska izmeklēšana pirms implantācijas.
Mutācijas VHL gēnā var būt pārmantotas vai izveidoties de novo (apmēram 20%). Jaunas mutācijas gadījumā diagnoze var tikt noteikta, ja pacientam ģimenes anamnēzē nav VHL slimības, bet ir:
- divas vai vairāk hemangioblastomas,
- hemangioblastoma un feohromocitoma,
- hemangioblastoma un gaiššūnu nefrokarcinoma,
- multipli ar VHL saistīti tumori,
- viens ar VHL saistīts tumors (
Somatiska mozaīcisma dēļ pacientiem reti var būt klīniskie VHL slimības simptomi bez atrodamas mutācijas. Mutācija notiek embrionālās attīstības laikā pēc fertilizācijas; tādējādi dažas šūnas būs normālas, bet pārējās būs mutācijas nēsātājas. Jo agrīnāk mutācija notiek, jo vairāk audu iesaistās. Hematopoētiskas cilmes šūnas nesatur mutāciju, tāpēc perifērajās asinīs VHL ģenētiskais tests ir negatīvs.
Uzraudzības protokols
VHL pacienta uzraudzības protokols dažādos vecumposmos apkopots tabulā.